

When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how does the FDA make sure it’s safe after it’s on the shelf? Unlike brand-name drugs, which go through years of clinical trials before approval, generic drugs are approved based on one key fact: they’re bioequivalent to the original. That means they deliver the same active ingredient at the same rate and amount. But bioequivalence doesn’t guarantee every batch will behave the same in every patient. That’s where post-approval surveillance kicks in.
What Happens After a Generic Drug Gets Approved?
The FDA doesn’t stop watching once a generic drug hits the market. In fact, the real work begins then. The Center for Drug Evaluation and Research’s Office of Generic Drugs (OGD) runs a dedicated team called the Clinical Safety Surveillance Staff (CSSS). This group of doctors, chemists, and data analysts tracks about 1.2 million adverse event reports every year. These reports come from doctors, pharmacists, patients, and manufacturers. Some are serious-like a heart rhythm problem after taking a generic blood thinner. Others are annoying but still important-like a patch falling off or a tablet not dissolving.The Hidden Signal: The Weber Effect
There’s a well-documented spike in reports when a new generic drug launches. It’s called the Weber Effect. Right after approval, doctors and patients start noticing and reporting issues-sometimes 300% to 400% more than before. Why? Because now there are multiple versions of the same drug on the market. People compare them. Pharmacists notice differences. Patients call in when their medication feels different. This isn’t necessarily a sign the drug is dangerous-it’s a sign people are paying attention. The FDA uses this surge as a trigger to dig deeper. They look at which manufacturer’s version is getting the most complaints. If one company’s tablets are causing 70% of the reports but only hold 30% of the market, that’s a red flag.Where the Data Comes From
The FDA doesn’t rely on guesswork. It pulls data from multiple sources:- Drug Quality Reporting System (DQRS): This tracks about 50,000 quality complaints a year-things like discolored pills, broken tablets, or liquid medications with visible particles.
- MedWatch: Over 140,000 reports a year come from healthcare providers and patients through this voluntary reporting system. Pharmacists make up 42% of these reports.
- Prescription sales data: Tools like IMS Smart and Symphony show how much of each generic version is being sold. This helps the FDA tell if a problem is real or just a fluke. If a drug with 15% market share has 60% of the complaints, it’s worth investigating.
- Medical literature: A team reviews more than 500 journals every month for case studies or new safety concerns.
- Active surveillance networks: The FDA uses the Sentinel Initiative, which links data from 19 healthcare systems covering over 100 million patients. This helps spot rare side effects that might not show up in smaller reports.

What They’re Looking For
Generic drug surveillance isn’t just about the active ingredient. It’s about the whole package. The FDA watches for:- Manufacturing flaws: A tablet that doesn’t dissolve properly, a capsule that leaks, or a patch that won’t stick.
- Excipient differences: Inactive ingredients like fillers or dyes can cause reactions in sensitive patients. One generic version might use a different binder that makes the pill harder to swallow.
- Therapeutic inequivalence: This is the trickiest one. A drug might be bioequivalent on paper, but in real life, it doesn’t work the same. For example, extended-release metformin from one manufacturer might wear off after 18 hours instead of 24, causing blood sugar spikes. This is hard to catch because it doesn’t show up as an adverse event-it shows up as a treatment failure.
- Complex delivery systems: Inhalers, patches, and injectables are harder to copy exactly. Between 2018 and 2022, 12% of all safety issues came from these types of products.
How the FDA Decides What to Do
Every month, a committee made up of experts from safety, quality, and surveillance teams meets to review signals. They don’t just look at numbers-they read the stories behind them. A single report might say, “My blood pressure spiked after switching generics.” Another might say, “All 12 tablets in this bottle were cracked.” Each one gets reviewed by a medical officer. If a pattern emerges, they do a Health Hazard Evaluation (HHE). This ranks the risk: is the problem likely to happen? How bad would it be? Mild? Severe? Then they decide: do we need a recall? A warning? A change in labeling? Between 2020 and 2022, CSSS completed about 130 HHEs a year. Most issues are resolved with the manufacturer fixing the process or changing the packaging. But sometimes, the FDA steps in with a public alert or even a mandatory recall.The Gaps in the System
The system works well for obvious quality problems-like broken pills or wrong colors. But it struggles with subtle differences in how a drug works over time. Dr. Robert Temple, a former top FDA official, admitted the system is “excellent for detecting quality defects but less sensitive to subtle efficacy differences.” Why? Because the FDA doesn’t require manufacturers to test bioequivalence again after approval. For drugs with a narrow therapeutic index-like warfarin, levothyroxine, or lithium-even a tiny change in absorption can be dangerous. A 2021 Government Accountability Office report found that only 65% of potential therapeutic inequivalence signals were fully investigated. In one case, 217 patient reports about inconsistent thyroid medication took 18 months to fully review. Doctors also don’t always understand how the system works. A 2018 survey found 63% of family physicians believed the FDA does routine post-market bioequivalence testing. They don’t. That misunderstanding can lead to mistrust in generics when patients report feeling worse after a switch.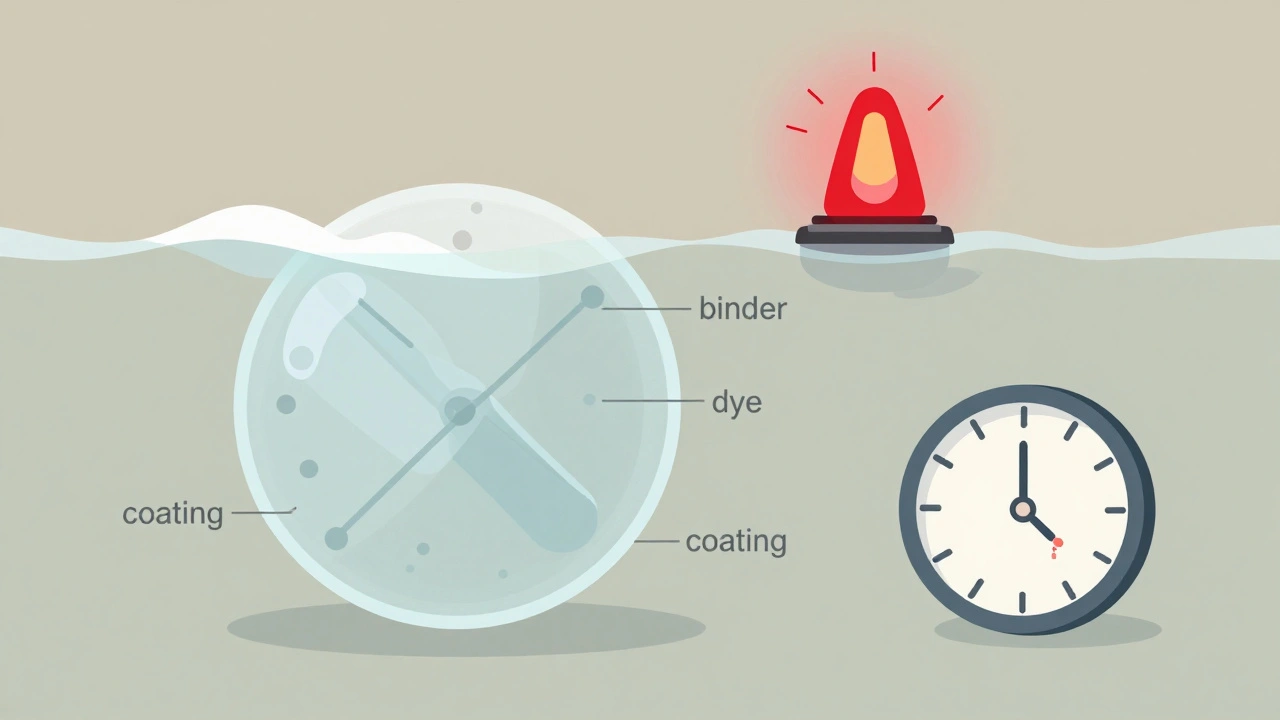
What’s Changing in 2025
The FDA is updating its approach:- AI is helping: New algorithms cut false positive signals by 27% in testing, so analysts aren’t chasing ghosts.
- Real-time data is coming: By late 2024, the FDA will start pulling pharmacy claims data directly-so they can see how many people are filling a generic and how often they refill it. A sudden drop in refills might mean patients are switching back to brand or stopping the drug entirely.
- Bioequivalence testing is getting stricter: Starting in 2025, the FDA plans to require post-approval bioequivalence studies for high-risk drugs like anticoagulants and seizure medications.
- Patient reporting is getting easier: A new portal for direct patient reporting on therapeutic inequivalence will launch in early 2025. Patients will be able to report if their medication “just doesn’t feel right”-even if they can’t name the side effect.
Why This Matters to You
If you take a generic drug, you’re part of this system. Your reports matter. If you notice your pill looks different, tastes odd, or doesn’t seem to work the same way, report it. Pharmacists report most of the time. But patients? Only 28% of those who submit reports through MedWatch ever get a follow-up. That’s changing. The FDA doesn’t monitor every batch. But it watches the patterns. And those patterns come from real people-doctors, pharmacists, and patients. The system isn’t perfect. But it’s built to catch problems before they become widespread. And with new tools and more transparency, it’s getting better.Does the FDA test generic drugs after they’re approved?
The FDA doesn’t routinely test every batch of generic drugs after approval. Instead, it monitors safety through reports from healthcare providers, patients, and manufacturers. If a pattern of problems emerges-like multiple reports of tablets not dissolving or blood levels being inconsistent-the FDA can request testing from the manufacturer or even initiate a recall. Post-approval bioequivalence testing is only required for certain high-risk drugs starting in 2025.
Can a generic drug be less effective than the brand-name version?
Legally, generics must be bioequivalent, meaning they deliver the same amount of active ingredient at the same rate. But in rare cases, differences in inactive ingredients or manufacturing processes can affect how the drug is absorbed over time-especially with extended-release or complex formulations. For drugs with a narrow therapeutic index, like levothyroxine or warfarin, even small differences can matter. The FDA has documented cases where patients experienced therapeutic failure after switching generics, even when the drug passed initial bioequivalence tests.
How do I report a problem with a generic drug?
You can report issues through the FDA’s MedWatch program. Visit fda.gov/medwatch or call 1-800-FDA-1088. Be specific: include the drug name, manufacturer, lot number (if available), what you experienced, and when it started. Even if you’re not sure it’s the drug’s fault, report it. The FDA uses these reports to spot patterns. Pharmacists and doctors can also report on your behalf.
Why do some people say their generic meds don’t work the same?
It’s not always in their head. Differences in fillers, coatings, or how the drug is released can affect how quickly it enters the bloodstream. For most people, this doesn’t matter. But for those on tight-dose medications-like thyroid hormones, seizure drugs, or blood thinners-these small changes can cause symptoms. The FDA tracks these reports and has seen cases where switching manufacturers led to clinical issues. If you notice a change after switching generics, talk to your doctor and consider reporting it.
Are generic drugs from foreign manufacturers less safe?
All generic drugs sold in the U.S., no matter where they’re made, must meet the same FDA standards. The FDA inspects foreign manufacturing sites just like U.S. ones. In fact, over 80% of generic drug ingredients come from outside the U.S., mostly from India and China. The FDA’s surveillance system tracks issues by manufacturer and lot number-not country of origin. Problems are usually tied to specific companies, not entire countries.
What to Do If You’re Concerned
If you’re worried about your generic medication:- Check the label for the manufacturer name and lot number.
- Don’t switch between different generic brands without talking to your pharmacist or doctor.
- Keep track of how you feel after switching. Write down any changes in symptoms, side effects, or effectiveness.
- Report anything unusual through MedWatch. Your report could help others.
- If you’re on a critical medication, ask your doctor if you should stay on the same generic manufacturer.
 Medications
Medications





Suzanne Johnston
December 7, 2025 AT 21:18The FDA's system is brilliant in theory but built on trust-and trust is fragile when you're the one swallowing the pill and feeling like something's off. It’s not about brand loyalty, it’s about bodily autonomy. If my thyroid meds suddenly make me feel like I’m moving through syrup, I don’t care if the bioequivalence numbers look good on paper. My body isn’t a statistical outlier-it’s my reality.
And yet, we’re told to just ‘report it’ like that fixes anything. The system doesn’t move fast enough for people who are suffering. Eighteen months to investigate 217 reports? That’s not surveillance-that’s negligence dressed up as bureaucracy.
They need real-time feedback loops, not just passive data collection. Patients aren’t just sources of noise-we’re the sensors. Stop treating us like background radiation and start listening like our lives depend on it. Because they do.
Graham Abbas
December 9, 2025 AT 15:27Man, this whole thing reminds me of how we treat mental health-until someone dies or explodes, nobody cares. The Weber Effect? That’s not a glitch, that’s a wake-up call. People are comparing pills like they’re comparing coffee brands. ‘This one makes me jittery,’ ‘That one gives me brain fog.’ And suddenly, the system wakes up.
But why wait for 400% spikes? Why not build in a baseline of patient-reported outcomes from day one? We’ve got apps, wearables, chatbots-why is the FDA still relying on handwritten forms and 1990s-era databases?
This isn’t about drugs. It’s about how we value lived experience over lab numbers. And until we fix that, we’re just playing whack-a-mole with side effects.
Elliot Barrett
December 10, 2025 AT 00:47Lol. So the FDA doesn’t test every batch? And you’re surprised people are skeptical? I’ve seen generics that look like they were made in a garage. One time my blood pressure med was a different shade of blue. I thought I was getting scammed.
Meanwhile, the brand-name stuff costs $300 a month. I get why people switch. But now I’m just waiting for the day someone dies because a tablet didn’t dissolve right and the FDA didn’t care until 300 people complained.
They’re not protecting us. They’re protecting profits. And we’re the guinea pigs.
Shubham Mathur
December 10, 2025 AT 01:58Bro the FDA is doing more than you think. I work in pharma in India and I know what goes into making these pills. The excipients matter. The coating matters. The humidity during packaging matters. But the system works if you report. I reported a batch that tasted metallic. Took 6 weeks. They recalled it. You think that’s nothing? That’s thousands of people spared from nausea and headaches.
Stop blaming the system. Use it. The MedWatch portal is free. Type it out. Send it. Be the change. Don’t just complain on Reddit. Do something.
Also stop saying ‘foreign = bad.’ My factory in Hyderabad makes 2 million pills a day for the US. FDA inspectors came last month. They cried. Not because we failed. Because we passed with flying colors. Stop the xenophobia.
And yes I used too many periods. I’m tired.
And yes I used too many periods. I’m tired.
And yes I used too many periods. I’m tired.
Ryan Brady
December 10, 2025 AT 09:05USA best! 🇺🇸 We got the best drugs in the world! Why are people even mad? The FDA is literally the gold standard! Other countries can’t even make pills that don’t fall apart! 😤
My cousin took a generic and said he felt ‘weird’-probably just stress or woke nonsense. Stop making everything a crisis. The system works. Trust the process. #FDAapproved #MadeInUSA
Delaine Kiara
December 10, 2025 AT 11:33Okay so let me get this straight-your thyroid medication can be ‘bioequivalent’ but still make you feel like you’re slowly turning into a zombie? And the FDA doesn’t test it again after approval? And you’re supposed to just… wait? For months? For a pattern to emerge?
And then when you report it, you get a robot email that says ‘Thank you for your feedback’? That’s not a system. That’s a joke. A really sad, bureaucratic, soul-crushing joke.
I switched from one generic to another last year and my anxiety went from ‘manageable’ to ‘I might need to move to a cabin in the woods.’ I didn’t even know it was the pill until I Googled ‘generic levothyroxine bad reactions’ and found 12,000 posts. Twelve thousand. And the FDA says ‘we’re working on it.’
Working on it? For five years? What does that even mean? That you’re writing a PowerPoint?
And now they’re adding AI? Great. So now a machine will decide if my panic attacks are ‘statistically significant’ enough to matter. I’m not a data point. I’m a person who can’t sleep because my heart won’t stop racing. And I’m supposed to be grateful because they’re ‘improving’?
They’re not improving. They’re just getting better at ignoring us.
Also, I’m starting a support group. If you’ve ever felt ‘off’ after switching generics, DM me. We’re not alone. And we’re not crazy. We’re just sick of being treated like ghosts.
Gilbert Lacasandile
December 11, 2025 AT 23:19I think the system’s actually pretty solid, all things considered. I’ve been on generics for over a decade-thyroid, blood pressure, cholesterol-and never had an issue. I know people have bad experiences, and I don’t minimize that. But the fact that the FDA tracks 1.2 million reports a year? That’s massive.
And the fact that they’re starting to require post-approval testing for high-risk drugs? That’s a huge step. Most people don’t realize how hard it is to replicate a drug’s delivery system. Patches, inhalers, extended-release tablets-they’re engineering nightmares.
It’s not perfect, but it’s not broken either. We just need to be better at reporting, and the FDA needs to be better at responding. Not with lawsuits or panic, but with clear communication. Patients just want to know: is this safe? And right now, the answer is usually yes. But we need to make sure it stays that way.
Haley P Law
December 12, 2025 AT 17:44OMG I just switched generics and my brain feels like mush. I swear I didn’t change anything else. I’ve been on this med for 8 years. Now I can’t focus. I’m crying for no reason. I think it’s the pill. I’m gonna report it. I’m gonna tag the FDA on Instagram. I’m gonna make a TikTok. I’m gonna call my senator. I’m gonna start a petition. I’m gonna cry again. I’m so tired. I just want to feel normal. Why is this so hard? 😭