When you pick up a prescription at the pharmacy, nine out of ten times it’s a generic drug. These copies of brand-name medications save Americans billions of dollars every year, but how do we know they are safe and effective? The answer lies in the rigorous regulatory authority of the FDA, which oversees the generic drug approval process through the Abbreviated New Drug Application (ANDA) pathway. This system ensures that generics are therapeutic equivalents to their brand-name counterparts without requiring manufacturers to repeat expensive clinical trials.
The Legal Foundation: Hatch-Waxman and the ANDA Pathway
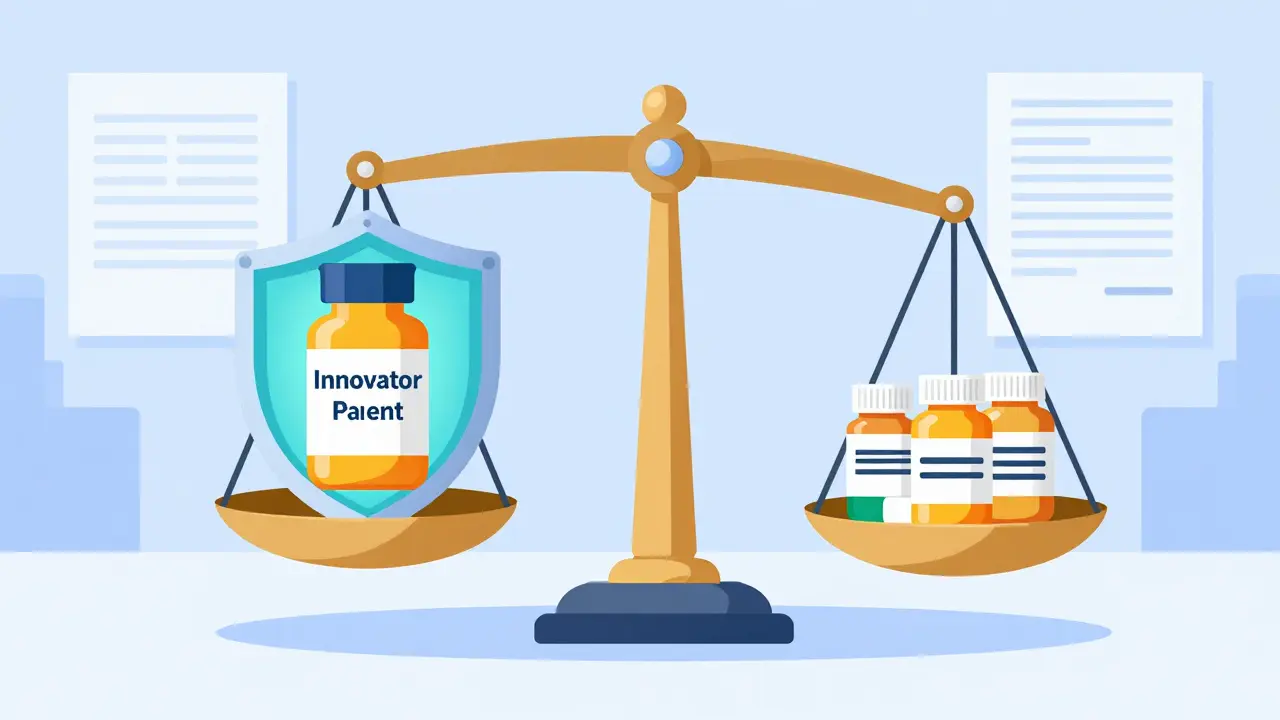
The current framework for approving generic drugs was established by the Hatch-Waxman Act of 1984. Officially known as the Drug Price Competition and Patent Term Restoration Act, this legislation created a balance between protecting innovator patents and encouraging competition. It introduced Section 505(j) of the Federal Food, Drug, and Cosmetic Act, which allows generic manufacturers to submit an Abbreviated New Drug Application (ANDA) instead of a full New Drug Application (NDA).
Unlike an NDA, which requires years of preclinical and clinical safety data, an ANDA relies on the existing safety profile of the reference listed drug (RLD). The generic manufacturer must only prove that their product is pharmaceutically equivalent and bioequivalent to the brand-name version. This streamlined approach reduces development costs from approximately $2.6 billion for new drugs to just $2.4-$6.3 million for generics, according to a 2020 RAND Corporation analysis.
What Manufacturers Must Prove: Bioequivalence Standards
To get approved, a generic drug must meet strict scientific criteria defined in 21 CFR 314.94. The FDA requires proof that the generic contains identical active ingredients, strength, dosage form, and route of administration as the brand-name drug. While inactive ingredients like dyes or fillers can differ, the core medicine must be the same.
The most critical test is bioequivalence testing. This involves pharmacokinetic studies measuring how quickly and how much of the drug enters the bloodstream. Typically, these studies use 24-36 healthy volunteers. For a generic to pass, the 90% confidence interval for the ratio of geometric means of Area Under the Curve (AUC) and maximum concentration (Cmax) must fall within 80.00%-125.00%. This narrow range ensures the generic works in the body just like the original.
- Identical Active Ingredient: Same chemical entity as the RLD.
- Same Strength: Identical milligram or unit dose.
- Same Dosage Form: Tablet, capsule, injection, etc.
- Bioequivalence: Rate and extent of absorption within 80-125% limits.
The Review Timeline: From Submission to Approval
The review process is managed by the Office of Generic Drugs (OGD) within the Center for Drug Evaluation and Research (CDER). Under the GDUFA III agreement, which started on October 1, 2022, the FDA has committed to specific Target Action Dates (TADs) to keep the pipeline moving.
Once submitted, an ANDA first undergoes a Filing Review. If the application is incomplete, it receives a Refuse-to-Receive (RTR) letter. In 2022, about 15.3% of submissions faced this hurdle, often due to missing chemistry, manufacturing, and controls (CMC) sections. If it passes filing, it moves to Substantive Review.
| Review Type | Target Action Date (TAD) | Application Fee (2022) |
|---|---|---|
| Standard ANDA | 10 months | $389,490 |
| Priority ANDA | 8 months | $389,490 |
| Facility Fee | N/A | $207,700 - $415,400 |
Priority reviews are reserved for drugs in shortage or first-time generics that break a brand-name monopoly. In fiscal year 2023, the FDA approved 1,256 ANDAs, a 12.7% increase from the previous year. However, challenges remain, with 14.8% of applications receiving Complete Response Letters due to bioequivalence study deficiencies.

Quality Control: CGMP and Facility Inspections
Approval isn’t just about the drug molecule; it’s about how it’s made. Manufacturers must comply with Current Good Manufacturing Practice (CGMP) regulations outlined in 21 CFR parts 210 and 211. These rules cover everything from raw material sourcing to final packaging.
The FDA inspects facilities to ensure compliance. According to the 2023 Generic Drug Program report, the inspection rate for generic drug facilities averaged 82.7% annually. Failures here are common reasons for RTR decisions, cited in 41.7% of refusal letters. With 78% of active pharmaceutical ingredients for generics coming from outside the U.S., supply chain resilience is a major focus for the agency.
Complex Generics: The New Frontier
Not all generics are simple tablets. Complex generics include inhalers, topical creams, extended-release formulations, and ophthalmic solutions. These products require specialized bioequivalence methodologies because standard blood tests don’t always reflect local delivery or complex release mechanisms.
In fiscal year 2023, 37.5% of ANDA approvals involved complex products, up from 22.1% in 2018. Dr. Sally Choe, Deputy Center Director for Pharmaceutical Quality at CDER, noted that the review process has evolved to address this complexity while maintaining scientific rigor. The FDA’s Complex Generic Drugs Initiative has helped improve approval rates for these difficult products, rising to 83.6% in 2023 from 67.2% in 2018.

Impact on Healthcare Costs and Patient Access
The primary goal of the ANDA system is affordability. Generics comprise 90% of prescriptions filled in the United States but account for only 23% of total spending. This generates roughly $132.6 billion in annual savings, according to the Generic Pharmaceutical Association’s 2023 Savings Report.
Pharmacists report that FDA-approved generics reduce patient out-of-pocket costs by 80-85% compared to brand-name equivalents. For example, the cost of insulin dropped significantly after the FDA approved biosimilars like Semglee. Despite these benefits, some patients express concern about efficacy. However, FDA investigations found that 92.3% of adverse event reports citing perceived differences were due to disease progression rather than the drug itself.
Future Outlook: GDUFA IV and AI Integration
Looking ahead, the FDA is modernizing its approach. Negotiations for GDUFA IV concluded in September 2024 with a $2.1 billion funding commitment through 2027. A significant portion, $412 million, is dedicated to complex generics development.
In October 2025, the FDA announced a pilot prioritization program for ANDA reviews, offering faster reviews for companies manufacturing in the U.S. Additionally, the agency is piloting artificial intelligence-assisted review for 12% of ANDAs to speed up processing. By 2027, analysts project the approval rate could reach 1,500-1,700 applications annually, driven by a backlog of over 2,100 pending first-generic applications.
How long does it take for the FDA to approve a generic drug?
Under GDUFA III, the target action date for a standard ANDA is 10 months from submission. Priority applications, such as those for drugs in shortage, have a target of 8 months. However, if an application is incomplete, it may receive a Refuse-to-Receive letter, delaying the process.
Are generic drugs as effective as brand-name drugs?
Yes. The FDA requires generics to demonstrate bioequivalence, meaning they deliver the same amount of active ingredient into the bloodstream in the same amount of time as the brand-name drug. Studies show 92.3% of reported issues with generics are related to disease progression, not product failure.
What is the difference between an NDA and an ANDA?
An NDA (New Drug Application) is used for new drugs and requires extensive clinical trial data to prove safety and efficacy. An ANDA (Abbreviated New Drug Application) is used for generics and relies on the existing safety data of the brand-name drug, requiring only proof of bioequivalence and manufacturing quality.
Why do some generic drugs look different from brand names?
Generic drugs can have different inactive ingredients, such as dyes, flavors, or binders. These do not affect the therapeutic effect of the drug. The active ingredient, strength, dosage form, and route of administration must be identical to the brand-name reference listed drug.
What is the Hatch-Waxman Act?
The Hatch-Waxman Act of 1984 created the legal framework for generic drug approval in the U.S. It balanced patent protection for innovator drugs with expedited pathways for generic competitors, introducing the ANDA process to lower healthcare costs.
What are complex generics?
Complex generics are drug products that present unique challenges in demonstrating bioequivalence, such as inhalers, topical creams, extended-release formulations, and eye drops. They require specialized testing methods beyond standard blood concentration studies.
 Medications
Medications