

Every pill, injection, or capsule that reaches a pharmacy shelf has passed through one final, non-negotiable gate: batch release testing. This isn’t just paperwork or a formality. It’s the last line of defense between a patient and a potentially dangerous product. If a batch fails here, it never leaves the facility. No second chances. No exceptions.
What Exactly Happens During Batch Release Testing?
Think of each batch of medicine as a unique product, even if it’s made using the same recipe. Raw materials vary slightly. Machines wear down. Environmental conditions shift. Batch release testing checks that every single batch meets the exact standards approved by regulators - no more, no less. For a small molecule drug like a common antibiotic tablet, testers look at:- Identity: Is this actually the drug it claims to be? Tests like HPLC or FTIR confirm the chemical structure matches the approved formula.
- Strength (Assay): Does each tablet contain 90-110% of the labeled dose? Too little? It won’t work. Too much? It could harm the patient.
- Purity (Impurities): Are there any unwanted byproducts? ICH guidelines limit unknown impurities to 0.10% in most new drugs. Even tiny amounts can cause side effects.
- Dissolution: Will the tablet break down properly in the body? For generics, the dissolution profile must match the original brand within an f2 similarity factor of 50 or higher.
- Physical properties: Tablet hardness (usually 4-10 kp), size, color, and coating consistency are visually and mechanically checked.
For injectables and biologics, the stakes are higher. Tests include:
- Endotoxin levels: Must be under 5.0 EU/kg/hr for spinal injections. A single contaminated vial can cause septic shock.
- Particulate matter: Small particles in IV solutions can block capillaries. Limits are strict: no more than 6,000 particles/mL ≥10μm.
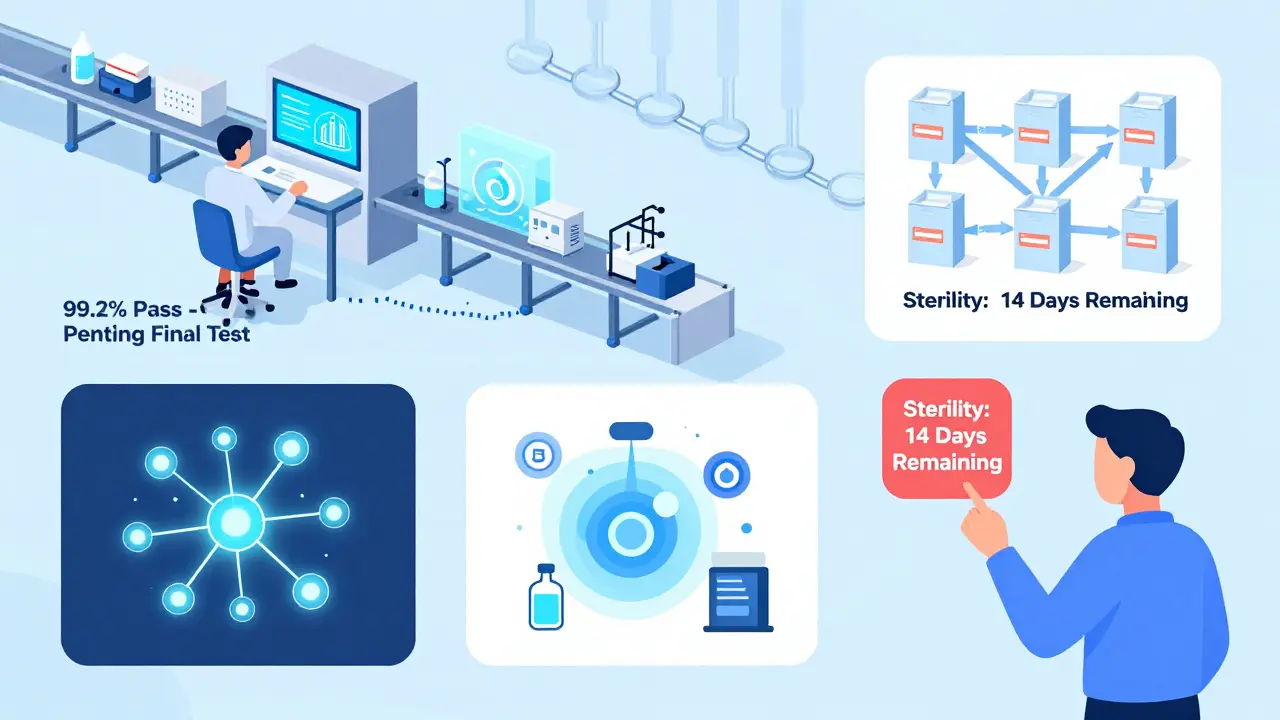
- Microbial limits: Non-sterile products can’t exceed 100 CFU/g. Sterile products? They must be completely free of microbes - tested via sterility assays that take 14 days to complete.
- Potency: Biologics like monoclonal antibodies require biological assays validated under USP <1033>. This isn’t a simple chemical reading - it’s a live-cell test measuring how well the drug triggers the intended immune response.
Stability testing runs in parallel. A sample from each batch is stored under accelerated conditions (40°C/75% RH) for six months and real-time conditions (25°C/60% RH) for up to three years. If the drug degrades too fast, the whole batch is scrapped.
Who Signs Off? The Qualified Person (QP) and the Quality Unit
Testing alone doesn’t release a batch. Someone has to review every single result, every instrument printout, every calculation. In the EU, that’s the Qualified Person - a certified professional with at least five years of GMP experience and formal training. They must sign off before the product leaves the plant. In the U.S., it’s the Quality Unit - a team of trained professionals who verify that the batch was made according to approved procedures and that all tests passed. Two analysts must independently review each critical test result. No single person has final authority.This isn’t just about checking boxes. It’s about accountability. In 2023, an FDA Form 483 cited a major manufacturer for releasing 12,000 vials of a monoclonal antibody with subpotent batches. The root cause? Inadequate review procedures. The result? A $9.2 million recall and an 18-month import ban.
Why Does This Cost So Much - and Take So Long?
A single batch of a complex biologic can take 21 to 35 days to fully test and release. Why? Because some tests can’t be rushed.- Sterility tests take 14 days - you can’t speed up microbial growth.
- Stability studies require months of storage before results are valid.
- Method validation - proving your test works reliably - can take 6-12 months for new products.
Documentation is another time sink. Every raw chromatogram, every instrument log, every analyst’s handwritten note must be retained for at least one year after the product expires - sometimes longer. In 2024, a senior QP in Germany reported spending 40-60 hours per batch just reviewing paperwork for biologics.
And it’s getting more expensive. Since 2020, testing costs have risen an average of 22% due to stricter limits, more complex products, and higher regulatory scrutiny. The global batch release testing market is now a $2.8 billion industry, growing at 6.7% per year.

What Goes Wrong? The Top 3 Reasons Batches Fail
Despite all the systems in place, batches still fail. According to the Parenteral Drug Association’s 2024 report, 83% of failures fall into three categories:- Dissolution testing (32%): The tablet doesn’t dissolve fast enough, or too fast. Often due to changes in excipients or manufacturing pressure.
- Impurity profiles (28%): Unexpected byproducts appear. Could be from a new supplier of raw material, or a change in reaction conditions.
- Microbial contamination (23%): Even a single breach in cleanroom protocols can ruin a batch. Airflow issues, improper gowning, or unsterilized equipment are common causes.
Other failures - identity mix-ups, potency shifts, particulate contamination - make up the remaining 17%. Most of these are preventable with better training, tighter process controls, and automated systems.
How Are Companies Making This Faster and More Reliable?
The industry is slowly moving away from waiting for lab results to release batches. Two big changes are gaining ground:1. Predictive Release Testing - Using sensors and real-time data from the production line (called Process Analytical Technology, or PAT), companies can now predict whether a batch will pass final tests before it’s even finished. The FDA’s 2025 pilot program allows this for continuous manufacturing facilities - but only 12 companies have qualified so far. The payoff? 34% fewer batch failures, according to PharmaIntelligence.
2. Digital Quality Systems - Manual reviews are slow and error-prone. Integrated Laboratory Information Management Systems (LIMS), like Thermo Fisher’s SampleManager, automate data collection, flag outliers, and streamline approvals. A 2024 AAPS survey found that companies using LIMS saw a 22% reduction in batch release time.
AI is also creeping in. Some firms use machine learning to spot patterns in historical data that predict failure risks. But regulators are cautious. EMA’s 2024 pilot showed AI was 78% accurate - but the FDA demands 99.9% confidence before accepting it as a replacement for traditional testing.
Regulatory Differences Around the World
What’s required in the U.S. isn’t always the same as in Europe or China.- EU: Every batch must be tested in full. No shortcuts. The Qualified Person must certify each one.
- U.S.: The FDA allows reduced testing for companies with proven process control - especially under continuous manufacturing. But they require real-time data and full traceability.
- China: Since 2023, imported vaccines must undergo mandatory batch release testing by NMPA before entry - adding 14-21 days to the supply chain.
This patchwork of rules makes global distribution complex. A batch that passes in the U.S. might get held up in Europe because of a minor difference in impurity limits or documentation format.
What’s Next? The Future of Batch Release
The future isn’t about eliminating batch testing - it’s about making it smarter.- ICH Q14 (effective Nov 2024): Lets companies design more flexible, risk-based testing plans. For stable, well-understood products, you might test less often.
- ICH Q2(R2) (planned 2026): Will introduce Quality by Design principles into test method selection - meaning tests are chosen based on how they impact patient safety, not just tradition.
- Blockchain traceability: The FDA is pushing for blockchain-based batch tracking by 2028. Every step - from raw material to patient - will be digitally recorded and immutable.
- Continuous quality verification: By 2030, 60% of advanced manufacturers may rely on real-time monitoring instead of end-of-line batch testing. But traditional testing will still dominate for legacy products through 2035.
One thing is certain: as drugs get more complex - especially biologics and gene therapies - batch release testing will only become more critical. The tools will change. The regulations will evolve. But the goal won’t: no unsafe medicine reaches a patient.
Is batch release testing required for all drugs?
Yes. Every batch of a marketed drug - whether it’s a simple aspirin tablet or a cutting-edge gene therapy - must pass batch release testing before distribution. This is mandated by regulators worldwide, including the FDA, EMA, and NMPA. Even over-the-counter products are subject to the same standards.
How long does batch release testing usually take?
It varies by product type. Small molecule generics take 7-10 days. Complex generics or injectables take 14-21 days. Biologics like monoclonal antibodies or vaccines can take 21-35 days, mainly due to sterility and potency tests that require incubation time. Stability testing runs for months but is done in parallel.
What happens if a batch fails release testing?
The batch is quarantined and rejected. It cannot be sold, distributed, or used. The manufacturer must investigate why it failed - whether it was a raw material issue, equipment malfunction, or human error. Depending on the cause, the batch may be reprocessed (if allowed), destroyed, or, in rare cases, retested after corrective actions. If the failure points to a systemic problem, regulators may issue a warning or recall other batches.
Can AI replace batch release testing entirely?
Not yet. AI can predict outcomes and flag risks, reducing the need for some physical tests - especially in continuous manufacturing. But regulators still require physical verification of identity, potency, purity, and sterility. AI is a tool to support decision-making, not replace the final lab-based certification. Full replacement would require 99.9% confidence, which no system has achieved yet.
Why is documentation so important in batch release?
Documentation proves the batch was made and tested correctly. Regulators don’t just check results - they check how you got them. Every instrument printout, calibration record, analyst signature, and calculation must be preserved for at least one year after expiration. Missing or altered records can lead to regulatory actions, recalls, or even criminal charges for fraud. Digital batch records are now mandatory for large manufacturers under the 2023 Drug Supply Chain Security Act.
 Medications
Medications



Mike Rose
January 30, 2026 AT 00:14lol so basically we pay $20 for a pill and $10k to test it? sounds like a scam.
Sidhanth SY
January 30, 2026 AT 05:02Interesting read. I work in pharma logistics in Bangalore and honestly, the delay in batch release is insane sometimes. One batch got stuck for 28 days just because the sterility test took its full 14 days and the QP was on vacation. No shortcuts, but man, it kills supply chains.
Sheila Garfield
January 30, 2026 AT 14:13It’s wild how much goes into something most people just swallow without a second thought. I used to think meds were just made and shipped. Now I see it’s like a 30-day audit before you even get a headache tablet. Respect.
April Allen
February 1, 2026 AT 00:28The shift toward ICH Q14 and risk-based testing is long overdue. For well-characterized small molecules with decades of stability data, full batch testing is redundant. We’re wasting millions on redundant assays while novel therapies languish in regulatory limbo. Quality by Design isn’t a buzzword-it’s the only scalable path forward.
Russ Kelemen
February 1, 2026 AT 03:35There’s something deeply human about this whole process. We’re not just testing chemicals-we’re testing our own accountability. Every signature on a logbook, every calibration record, every 14-day wait for sterility results-it’s all a promise. A promise that someone, somewhere, won’t get sick because we were careless. That’s not just compliance. That’s ethics in action.
Claire Wiltshire
February 2, 2026 AT 19:35For anyone unfamiliar with GMP: the Qualified Person (QP) role in the EU is non-negotiable. They’re legally liable for every batch released. That’s why they spend 50+ hours reviewing paperwork-because if they miss a single outlier, and someone dies, they could face criminal charges. This isn’t corporate bureaucracy. It’s legal and moral responsibility encoded into law.
Shawn Peck
February 4, 2026 AT 03:14AI is gonna replace all this in 5 years. You think they really need humans to stare at chromatograms? Nah. Machines are faster, cheaper, and don’t take coffee breaks. The FDA’s being stubborn. 99.9% confidence? We got self-driving cars that kill people and they’re on the road. Let AI do the work!
Adarsh Uttral
February 5, 2026 AT 00:43bro the part about particulates in IVs… i had a cousin get a blood clot from a contaminated bag. they never found out why. that 6,000 particles per ml limit? feels like a lottery. hope they fix this.